
SINGEK BLOG: Filling the Gaps with dark matter – reconstructing the tree of life
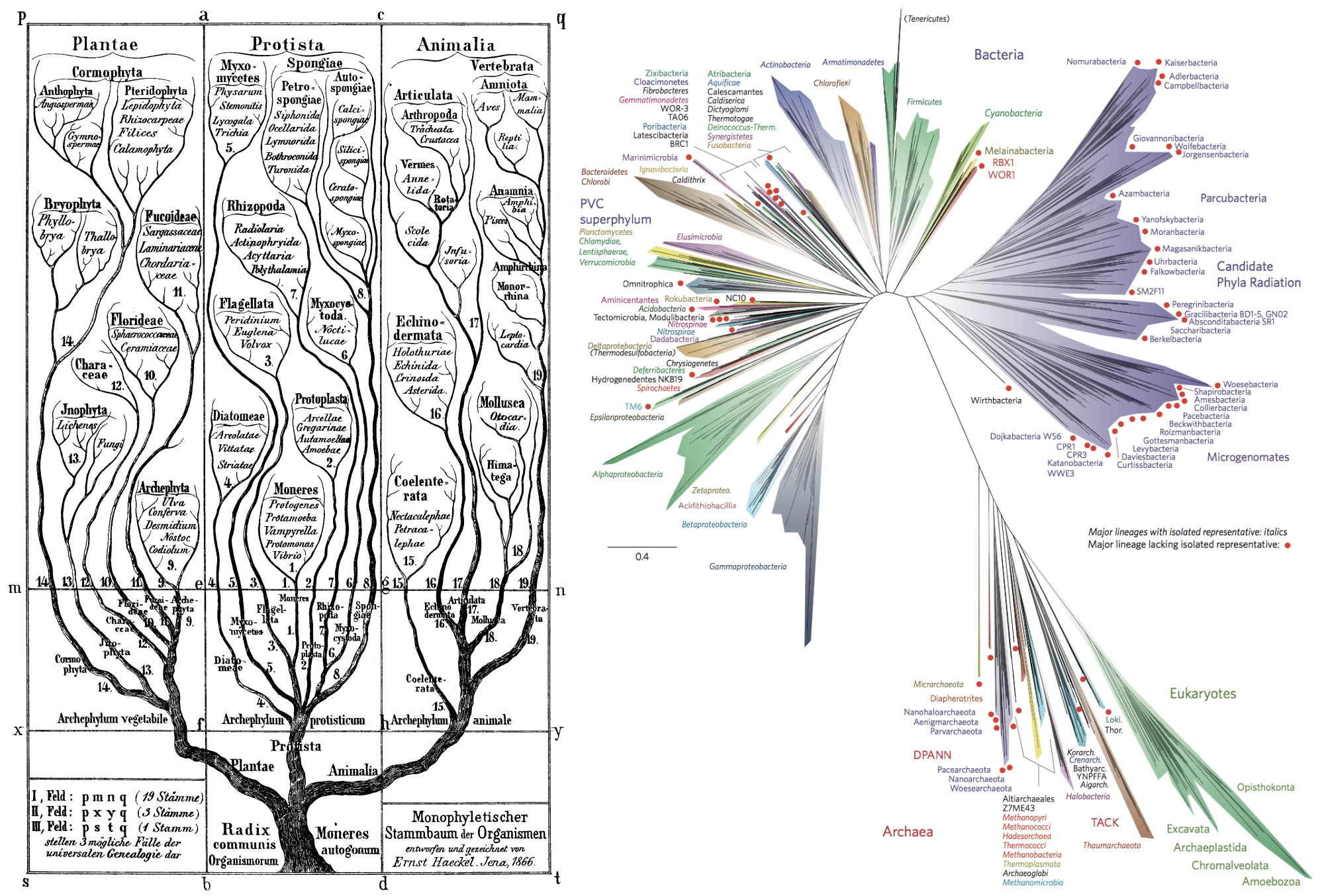
The tree of life as the scientist/artist Ernst Haeckel envisioned it in the late 19th century (bacteria and archaea were not known then as such); right: A current update of the tree of life using available genome sequences and covering a wide range of prokaryotes and several eukaryotic lineages.
The tree of Life as we currently display it, is full of gaps and missing parts, reflecting enormous evolutionary distances between organisms. These gaps make it hard to confidently make statements about their evolution. Luckily, we are living in exciting times, where environmental sequencing enables researchers to describe new orders and even phyla on a regular basis. It feels like a renaissance after the classical naturalists of the 18th and 19th century described many of the species known today. In the last year or two, several studies where published that exhibit an unprecedented magnitude of newly discovered organisms. Each of them presented hundreds or even thousands of complete genomes extracted from metagenomics sequences. Metagenomics means sequencing DNA from all the organisms present in a sample at the same time.
This so-called ‘microbial dark matter’ (because we did not know it existed before genome sequencing) can be used to fill the gaps in the tree of life. This can reduce the artifacts of phylogenetic inferences, which is the reconstruction of the evolutionary history between organisms from their genes and genomes. Our job is to make sure that the phylogenetic models we use integrate this huge number of newly discovered genomes. We are employing better models of sequence evolution but also integrate, for example models of protein family evolution or the evolution of protein structures. The results of this combined approach should be of greatest importance to the whole field of evolutionary biology. It will enable us to better understand microbial diversity and even shed light on parts of evolution that remain some of the greatest mysteries in biology, such as the origin of eukaryotes.
References
Haeckel, E. Generelle Morphologie der Organismen (1866); Hug, L. A. et al. A new view of the tree of life, Nature Microbiol. 1, 16048 (2016).
About the author

Max Emil Schön – ESR 5
After my undergraduate degree in geoecology, and especially after a Bachelor’s project on the phylogenetic relationships and diversity of fungi I choose to change my field of studies to bioinformatics and computational biology. I wanted to be able to thoroughly understand the tools I was using and develop improvements or new tools by myself. Nevertheless, as I was mainly interested in those methods as a way to understand relationships between organisms (from an evolutionary as well as from an ecological perspective), I did not want to abandon geoecology neither. At the University of Tübingen I’ve had the awesome possibility to study both degrees in parallel, and I learned a lot about bioinformatics as well as evolution and biodiversity during this time. The background from both my studies was applied during a joint Master’s project, where I evaluated bioinformatics methods for the assessment of species diversity in environmental DNA samples using metabarcoding and applied those methods to a next-generation-sequencing dataset generated from symbiotic fungal communities in roots and soil.

Leave a Reply
Want to join the discussion?Feel free to contribute!